CSD Metal-Organic Framework Collection in Action: Toyota Motor Europe. Improving Hydrogen Physisorption Energy of MOFs for Energy Storage
Here we highlight a paper by researchers at the Material Engineering Division of Toyota Motor Europe and the University of Crete who used CCDC’s metal-organic framework (MOF) collection to investigate how ligand functionalization affects the hydrogen storage profile of MOFs. This is part of our series highlighting examples of the Cambridge Crystallographic Data Centre (CCDC) tools in action by scientists around the world.
Summary
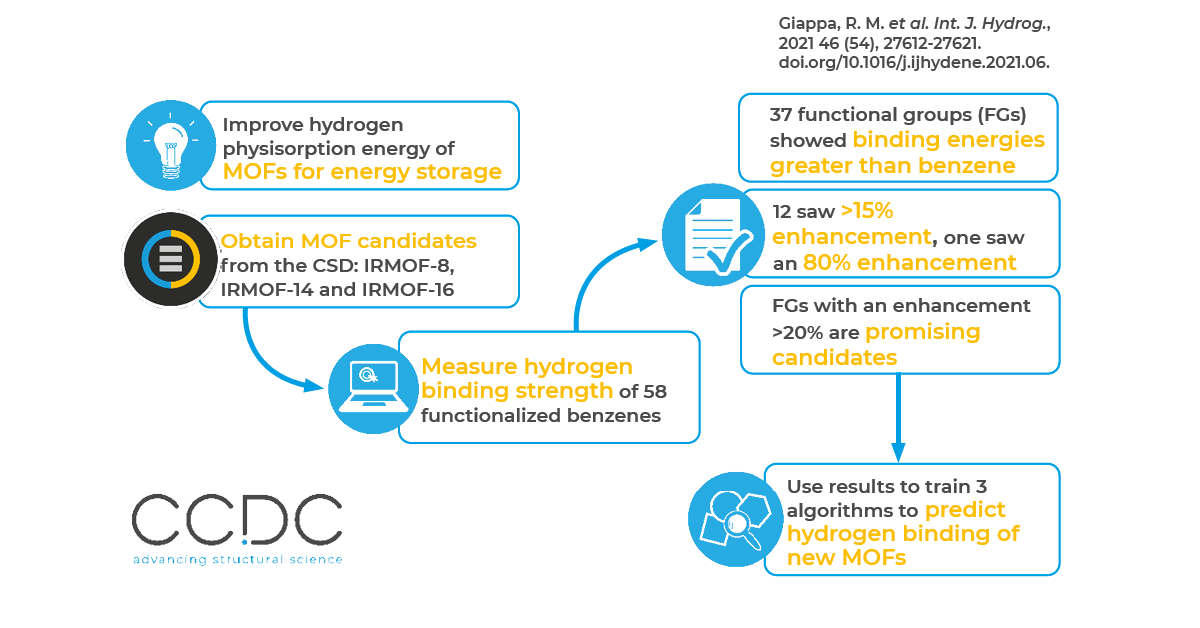
In this work, researchers combined multi-scale calculations with machine learning (ML), to investigate how ligand functionalization affects the hydrogen storage profile of MOFs. The functionalization of organic linkers can work well as a way to increase the H2 physisorption energy of MOFs. Aromatic backbones, like benzene and naphthalene, serve as organic linkers for many MOFs. The authors chose to focus on substituted benzene rings. They obtained the positions of the MOF framework atoms from the CSD and calculated the binding energy of hydrogen with 58 strategically selected functionalized benzenes using quantum chemistry (QM) computational methods. They found that many functional groups (including -OPO3H2, –OCONH2) increase hydrogen interaction up to 15 to 25% compared to benzene, while –OSO3H saw up to an 80% enhancement.
The researchers then completed a proof-of-principle ML analysis, which showed a good prediction of the H2 binding energies, even with their limited amount of data. From this, they concluded that their functionalization strategy can be applied to various porous materials in order to enhance their hydrogen storage performance.
Why
The researchers needed reliable structures on which to base their quantum computational and ML models. The Cambridge Structural Database (CSD) currently houses over 100,000 expertly curated MOF-like frameworks. With the structures in the CSD, this study was able to help explain how functional group composition affects hydrogen uptake profile, which is key to designing MOFs for storage tanks. Volumetric and gravimetric hydrogen capacities determine the size and weight of MOF-filled tanks that store hydrogen. These factors must be optimized so a storage tank is neither too large nor too heavy for reasonable use.
The researchers then used those findings as a database for an ML analysis, which can serve as an alternative to costly QM calculations. The ML results offered a fair prediction of the hydrogen binding energies and confidently identified low-lying structures in the interaction energy landscape. This approach can decrease the need for expensive computational efforts and extrapolate valuable physical-chemical insight
How
In order to investigate how linker functionalization impacts hydrogen adsorption, the researchers functionalized three isoreticular MOF candidates obtained from the CSD: IRMOF-8, IRMOF-14 and IRMOF-16. They then performed classical Monte Carlo simulations in the Grand Canonical ensemble (GCMC). They specifically studied the hydrogen uptake profile of both the parent structures and those functionalized with some of the best candidates. The different organic linkers were doubly functionalized.
Read More
Read the full paper: A combination of multi-scale calculations with machine learning for investigating hydrogen storage in metal organic frameworks, International Journal of Hydrogen Energy from Int. J. Hydrog., 2021.
Learn more about the CSD MOF collection, which contains over 10,000 MOF crystal structures available at no cost to academic researchers.
Learn more about the complete CSD, which currently houses over 100,000 MOF-like frameworks.
Explore more examples of CCDC tools in action.