Ligand Based Virtual Screening With CSD-Discovery
This blog is based on the videos from Abhik Mukhopadhyay, former Research and Application Scientist at the CCDC, who provided a brief introduction on what virtual screening is and how to do ligand based virtual screening using CSD-Discovery tools.
What Is Virtual Screening?
Virtual screening is a collection of several in-silico techniques used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme. Virtual screening can be used to select compounds for screening from in-house databases, to choose compounds to purchase from external suppliers, or to decide which compounds to synthesize next.
Virtual Screening Methods
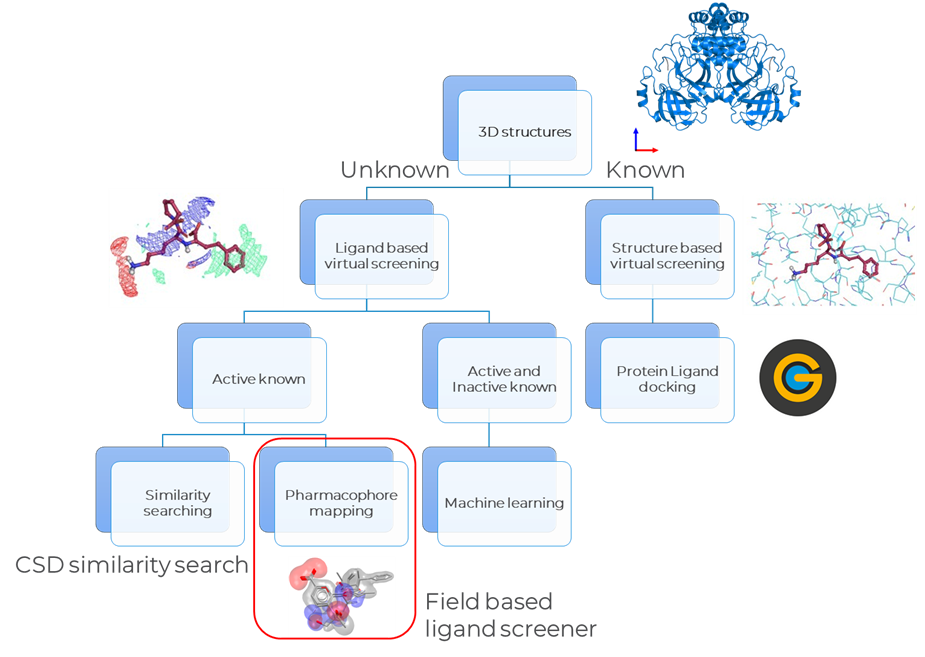
The technique applied while doing virtual screening depends mainly on the amount of information available about the particular disease target (Figure 1). If the 3D structure of the target is known, then the best method is the structure based virtual screening, which consists in protein–ligand docking. CSD-Discovery has a well validated program for this purpose: GOLD.
If instead the 3D structure of the target is not known, then the ligand based method is the first choice. Ligand based screening methods are more robust than docking into homology models or apo structures. They can also vary according on the type of information that is available. When the actives are known, the 2D similarity searching method can be used. CSD-Discovery includes a similar research method available through the CSD Python API tool. Alternatively, there is the pharmacophore mapping method, where a CSD Field-Based Ligand Screener performs this analysis. On the other hand, if the actives and inactives are both known, then the preferred method for virtual screening is machine learning.
How To Do Ligand Based Virtual Screening With CSD Tools
An overview of the CCDC tools that can be used to perform ligand based virtual screening are now described.
The first tool is the CSD Conformer Generator, which produces realistic ensembles of low energy ligand structures. These are ready to be exploited for drug design in the presence or in the absence of detailed knowledge about the three-dimensional structure of the protein active site. The second tool is the CSD Ligand Overlay, which overlays sets of flexible molecules that are assumed to be ligands of the same protein. The final tool we discuss is the CSD Field-Based Ligand Screener, which is available through the CSD Python API. This is a module that can be used to screen a library of compounds against a pharmacophore query obtained from one or multiple overlaid ligands.
CSD Conformer Generator
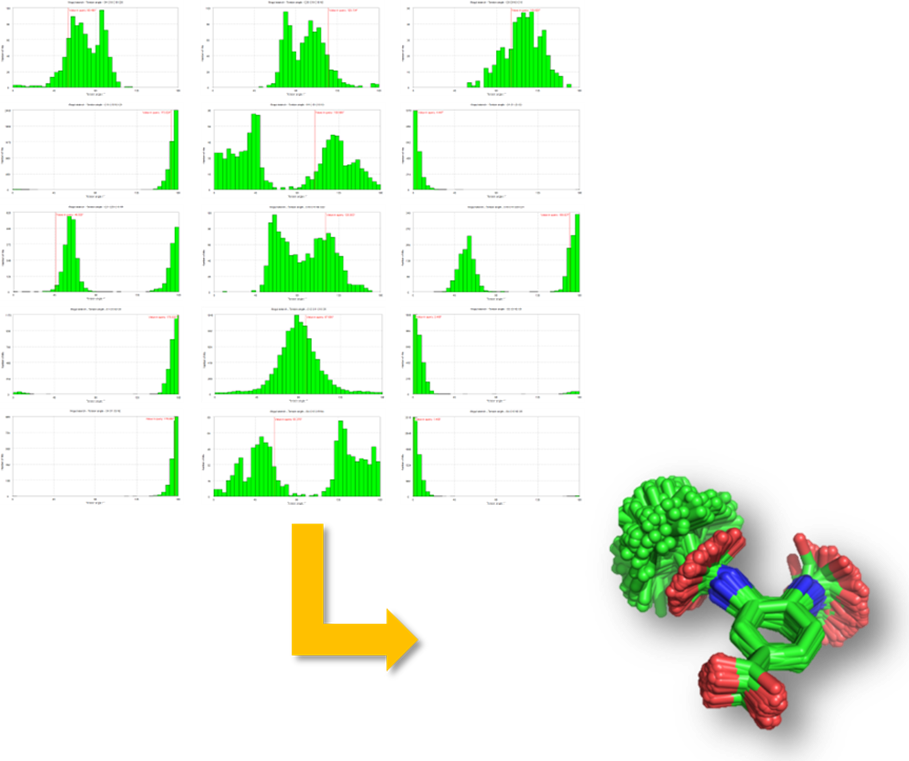
The CSD Conformer Generator is a knowledge-based program that uses the wealth of knowledge in the CSD to explore the conformational space of small molecules and generate plausible conformations of a small molecule (Figure 2). This tool provides the ability to both minimize molecular conformations and also generate diverse conformer subsets based on CSD data. Given a molecule in 3D, the bond lengths, angles, and dihedrals are minimized based on average values taken from Mogul, and the rotamers and ring distributions are assigned. The new version has improved handling of flexible ring systems. It is fast and useful, not only to generate plausible conformations of specific compounds, but also as optimization step to prepare compounds for virtual screening.
The CSD Conformer Generator is available as standalone executable, to be run on the command line. It is also available through the CSD Python API; through the Mercury interface, if looking at small molecule structures; through a script in the Hermes interface, if looking at small molecule structures within the context of a protein.
This tool has been evaluated extensively against the Platinum data set. More information on this specific aspect can be found in the article ‘Knowledge-Based Conformer Generation Using the Cambridge Structural Database’. [1]
CSD Ligand Overlay
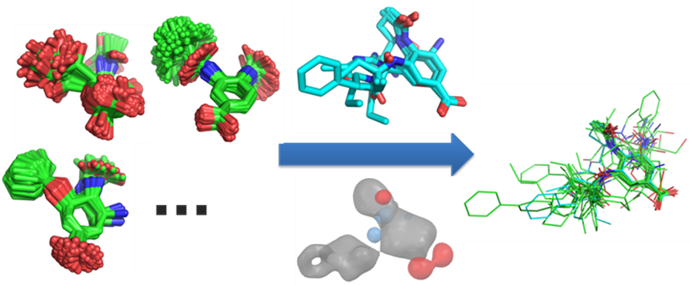
The purpose of the CSD Ligand Overlay software is to overlay sets of flexible molecules that are assumed to be ligands of the same protein sharing the same binding site (Figure 3). A correct prediction places the ligands in the conformations that they adopt when they bind to the protein. However, the software works in the absence of any protein-structure information, hence it is rarely possible to identify the correct overlay unambiguously. A variety of credible solutions are therefore produced.
The program represents each ligand by “fitting points”, which are specific features, i.e. chemical groups with particular physicochemical properties. Three types of features are always used: hydrophobic centroids, H-bond donors, and acceptors. In addition, customized features can be defined, e.g. positive or negative centres.
This overlay program has been validated extensively using the AZ test set, which contains 121 molecular overlays. More information on this specific aspect can be found in the literature. [2][3][4]
Want to try it for yourself? Follow the step-by-step tutorial on Ligand Overlay here.
CSD Field-Based Ligand Screener
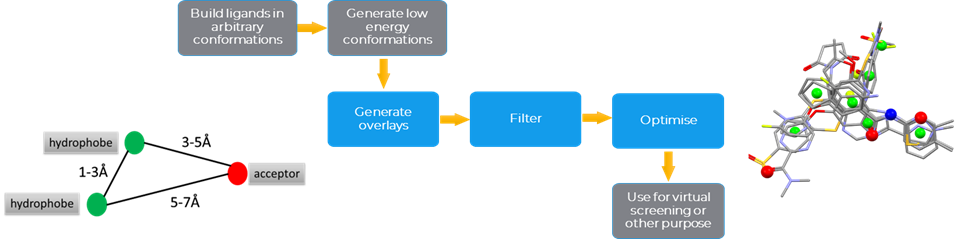
The CSD Field-Based Ligand Screener is available through the CSD Python API, as previously mentioned. It generates a field potential from a molecular overlay based on user-defined atom-types and creates fitting points in the hot-spots (Figure 4). It requires two input files (the query and the molecules to screen) and it globally optimizes the translation and rotation of each ligand by generating conformer libraries and then fitting the ligand atoms to precalculated fitting points. Finally, it screens the ligand conformational ensembles against the overlay query and ranks each of the screened ligands.
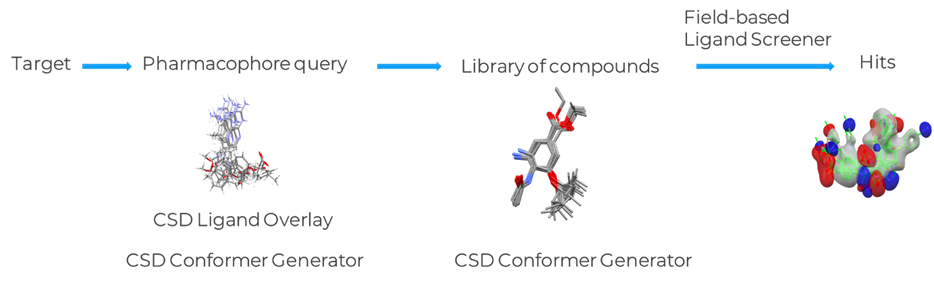
Ligand Based Virtual Screening Workflow
In this section, we show how the CSD-Discovery ligand screening tools can be used in a ligand based virtual screening workflow (Figure 5).
In the first step, ensembles of conformers are generated for a set of known inhibitors using the CSD Conformer Generator. Overlay hypotheses for these ligands are then produced using the CSD Ligand Overlay program and a pharmacophore model is selected for next step. The CSD Conformer Generator can also be used to generate a conformation of the library of active and inactive compounds before or during the screening step. Finally, a library of active and inactive compounds can be screened against the selected overlay solutions using the CSD Field-Based Ligand Screener, and the enrichment metrics are calculated.
A full tutorial can be found here, showing how these tools can be used in a real-life scenario, and how to calculate all the necessary metrics with the CSD Python API.
Next Steps
Want to try it for yourself? Follow the step-by-step tutorial on Ligand Based Virtual Screening here.
Watch the video – Drug Discovery: What’s the difference between structure based and ligand based virtual screening?
Watch the video – Create a ligand based screening workflow with CSD-Discovery.
Learn more about CSD-Discovery, GOLD, CSD Python API, Mogul, Mercury, Hermes.
To discuss further and/or request a demo with one of our scientists, please contact us via this form or .
References
[1] Cole J. C., et al. J. Chem. Inf. Model. (2018) 58, 615-629. https://pubs.acs.org/doi/10.1021/acs.jcim.7b00697
[2] Taylor R., et al. J. Comput. Aided. Mol. Des. (2012) 26, 451-472. https://link.springer.com/article/10.1007/s10822-012-9573-y
[3] Giangreco I., et al. J. Chem. Inf. Model. (2014) 54, 3091-3098. https://pubs.acs.org/doi/abs/10.1021/ci500509y
[4] Giangreco I., et al. J. Chem. Inf. Model. (2013) 53, 852-866. https://pubs.acs.org/doi/abs/10.1021/ci400020a