CSD Linker Database
Scaffold hopping is the replacement of part of a drug molecule with another, whilst retaining the features of the molecule most important for protein binding in their proper geometrical arrangement to interact with the receptor. This technique, particularly when carried out computationally on a protein-ligand crystal structure, is very popular with drug designers. It is used to develop molecules with more desirable physical properties, which escape existing patent coverage, or which may be easier to synthesise.
Download MOE CSD Linker Database
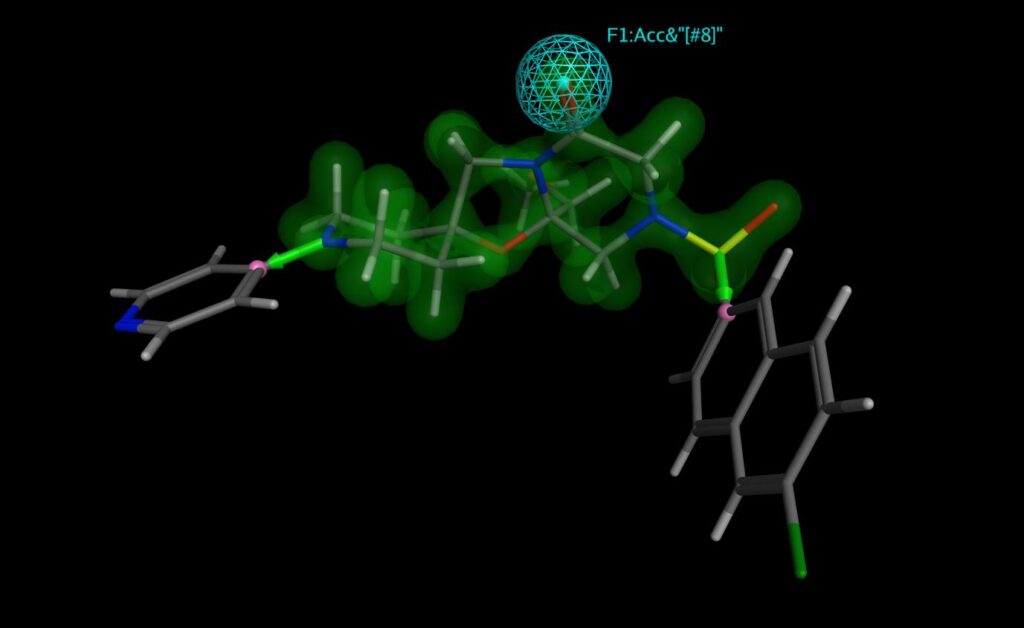
In Fragment-Based Drug Design, initial hit fragments are elaborated by adding chemical functionality to increase binding energy and efficiency (perhaps by addressing additional binding opportunities in a receptor pocket).
The Chemical Computing Group’s CSD Linker Database can be used in MOE’s Scaffold Replacement and Fragment Growing applications, along with judiciously chosen pharmacophore features, to speedily find new leads where the geometry of the linking or added fragments is low strain and the proposed compounds are very likely to be synthesisable.
A valid CSDS license is required for use of this database, which is provided in MOE-readable .mdb format only.
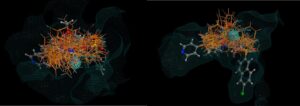
Figure 2: Two views of 26 hits (there were around 400 in all) found using the CSD linker database, superposed over the original molecule with the receptor surface highlighted. Note how none of the hits intrude into the space occupied by the receptor. No minimisation of each hit was carried out in this case to optimise the binding, but this is recommended.
